


 Copy to clipboard
Copy to clipboard 
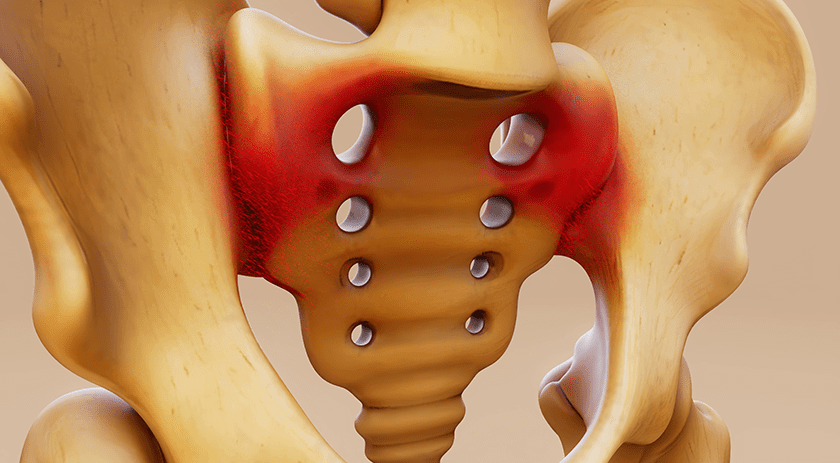
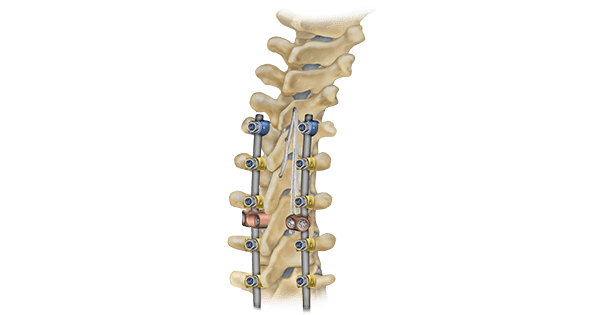
Medtronic received FDA 510(k) clearance and Breakthrough Device designation for its LigaPASS™ 2.0 Ligament Augmentation System. LigaPASS is reported to be the first and only FDA-cleared device with an indication for ligament augmentation in spine surgery.
Ligament augmentation has been studied for its impact on proximal junctional kyphosis (PJK), a frequent post-operative complication of adult spinal deformity surgery. While the exact cause is unknown, PJK is thought to involve disruption of spinal ligaments and can affect up to 46% of patients. The condition can significantly impact a patient’s quality of life. The severity of PJK can vary, and some patients may develop a more severe form of the condition called proximal junctional failure (PJF). Patients who develop PJF may develop structural and neurological complications, which increases the need for revision surgery.
A study of 242 adult spinal deformity cases found patients treated with ligament augmentation had significantly lower PJK and PJF complication rate as follows. After one year, the reoperation rate for adult spinal deformity patients treated with ligament augmentation was just 3.3% compared to 15.6% of not treated with ligament augmentation.
The ligament augmentation technique aims to reduce the reoperation rate for PJF and its associated costs, which can be more than $50,000 per procedure.
LigaPASS 2.0 can also be paired with the UNiD™ Adaptive Spine Intelligence (ASI) platform. LigaPASS 2.0 system connectors and bands may also be used in conjunction with the CD Horizon™ Solera™ spinal system.
“This clearance and Breakthrough Device designation demonstrates our ongoing commitment to innovation in spine surgery and delivering industry-leading solutions that improve care for patients and improve the experience for surgeons,” said Dan Wolf, vice president and general manager, intelligent Data Solutions at Medtronic.
Source: Medtronic
Medtronic received FDA 510(k) clearance and Breakthrough Device designation for its LigaPASS™ 2.0 Ligament Augmentation System. LigaPASS is reported to be the first and only FDA-cleared device with an indication for ligament augmentation in spine surgery.
Ligament augmentation has been studied for its impact on proximal junctional kyphosis...
Medtronic received FDA 510(k) clearance and Breakthrough Device designation for its LigaPASS™ 2.0 Ligament Augmentation System. LigaPASS is reported to be the first and only FDA-cleared device with an indication for ligament augmentation in spine surgery.
Ligament augmentation has been studied for its impact on proximal junctional kyphosis (PJK), a frequent post-operative complication of adult spinal deformity surgery. While the exact cause is unknown, PJK is thought to involve disruption of spinal ligaments and can affect up to 46% of patients. The condition can significantly impact a patient’s quality of life. The severity of PJK can vary, and some patients may develop a more severe form of the condition called proximal junctional failure (PJF). Patients who develop PJF may develop structural and neurological complications, which increases the need for revision surgery.
A study of 242 adult spinal deformity cases found patients treated with ligament augmentation had significantly lower PJK and PJF complication rate as follows. After one year, the reoperation rate for adult spinal deformity patients treated with ligament augmentation was just 3.3% compared to 15.6% of not treated with ligament augmentation.
The ligament augmentation technique aims to reduce the reoperation rate for PJF and its associated costs, which can be more than $50,000 per procedure.
LigaPASS 2.0 can also be paired with the UNiD™ Adaptive Spine Intelligence (ASI) platform. LigaPASS 2.0 system connectors and bands may also be used in conjunction with the CD Horizon™ Solera™ spinal system.
“This clearance and Breakthrough Device designation demonstrates our ongoing commitment to innovation in spine surgery and delivering industry-leading solutions that improve care for patients and improve the experience for surgeons,” said Dan Wolf, vice president and general manager, intelligent Data Solutions at Medtronic.
Source: Medtronic

You’ve reached your limit.
We’re glad you’re finding value in our content — and we’d love for you to keep going.
Subscribe now for unlimited access to orthopedic business intelligence.

JV
Julie Vetalice is ORTHOWORLD's Editorial Assistant. She has covered the orthopedic industry for over 20 years, having joined the company in 1999.